
El Origen de la Síntesis de Proteínas: Un Enigma Central en la Abiogénesis
El Paradigma del «Huevo y la Gallina» en la Biología Molecular
En el núcleo funcional de toda la vida conocida reside una interdependencia profunda y aparentemente irreductible entre los ácidos nucleicos y las proteínas. Los ácidos nucleicos, el ADN y el ARN, son los depositarios de la información genética, el manual de instrucciones que dicta la forma y función de un organismo.1 Las proteínas, por su parte, son las «máquinas» moleculares que ejecutan estas instrucciones; actúan como catalizadores (enzimas), componentes estructurales y sistemas de señalización que llevan a cabo la gran mayoría de los procesos celulares.1 Esta simbiosis funcional crea uno de los enigmas más profundos en el estudio del origen de la vida, a menudo denominado el problema del «huevo y la gallina».2
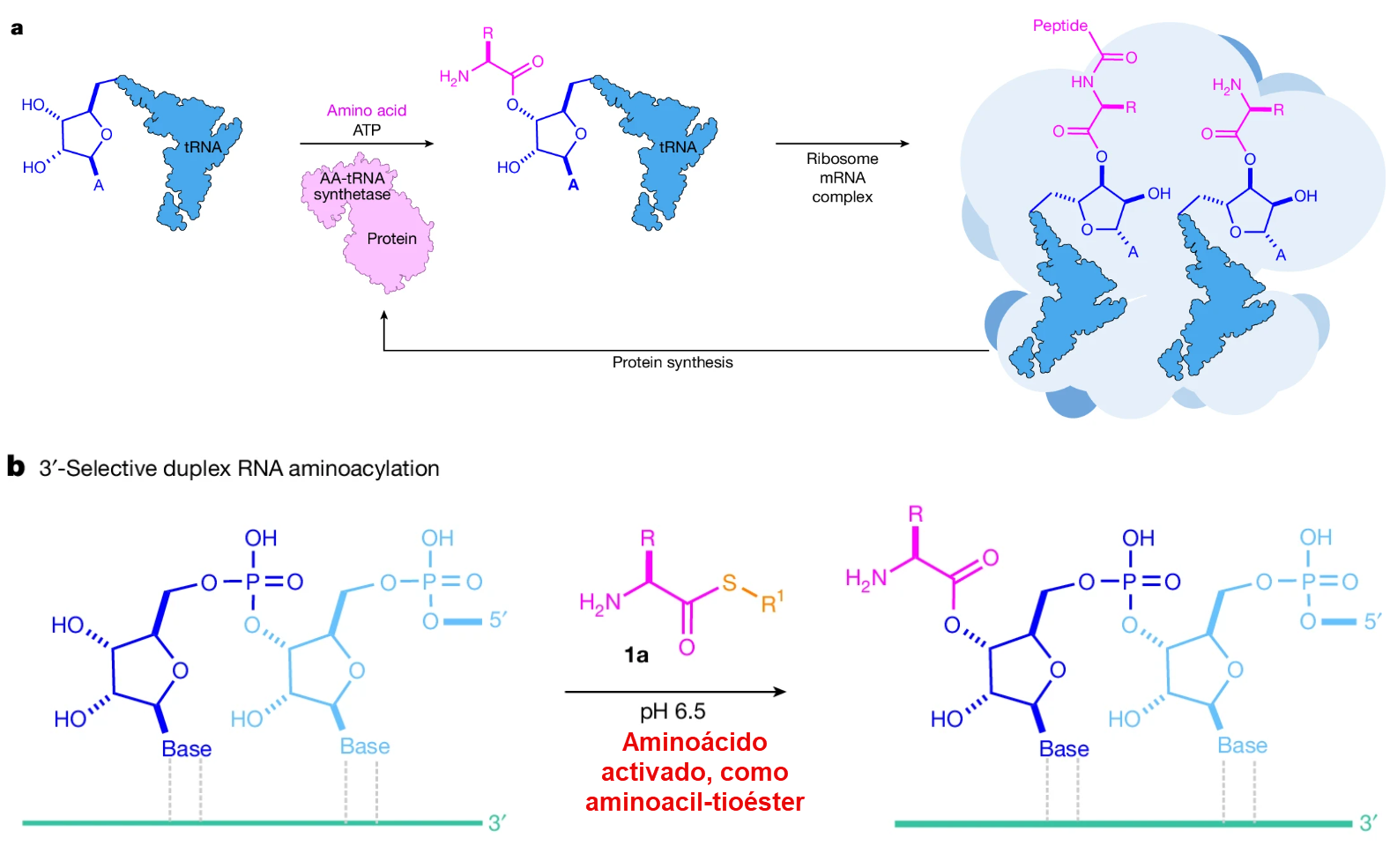
La maquinaria biológica contemporánea para la síntesis de proteínas, o traducción, es un sistema de una complejidad asombrosa. En su centro se encuentra el ribosoma, una enorme máquina molecular compuesta de ARN ribosómico (ARNr) y proteínas, que cataliza la formación de enlaces peptídicos.3 El proceso comienza cuando la información codificada en un gen de ADN se transcribe en una molécula de ARN mensajero (ARNm). El ribosoma se ensambla sobre este ARNm y lee su secuencia de nucleótidos en grupos de tres, conocidos como codones.3 Cada codón especifica un aminoácido particular. El papel de adaptador molecular lo desempeñan las moléculas de ARN de transferencia (ARNt), cada una de las cuales reconoce un codón específico a través de su bucle anticodón y lleva covalentemente unido el aminoácido correspondiente en su extremo 3′.3
El paso crucial que une el lenguaje de los nucleótidos con el de los aminoácidos es la aminoacilación, el proceso de «cargar» una molécula de ARNt con su aminoácido correcto.2 En la biología moderna, esta tarea es realizada con una fidelidad exquisita por una familia de enzimas proteicas llamadas aminoacil-ARNt sintetasas (aaRSs).2 Cada aaRS es específica para un aminoácido y su conjunto correspondiente de ARNt. Utilizando la energía de la hidrólisis del ATP, la aaRS activa el aminoácido y lo une covalentemente al extremo 3′ del ARNt.2 El ARNt cargado, o aminoacil-ARNt, está entonces listo para ser entregado al ribosoma.
Aquí radica la paradoja: la síntesis de proteínas (incluidas las propias aaRSs y las proteínas ribosómicas) requiere un conjunto preexistente de proteínas altamente especializadas. Sin embargo, estas proteínas no pueden existir sin un sistema de traducción que las fabrique. ¿Cómo pudo surgir un sistema tan intrincadamente interdependiente a partir de la simple química de la Tierra primitiva? Cualquier modelo plausible sobre el origen de la vida debe proporcionar una explicación de cómo se estableció este ciclo de retroalimentación positiva, superando la necesidad de una maquinaria proteica compleja en sus etapas iniciales.
Hipótesis en Competencia: El «Mundo de ARN» y el «Mundo de Tioésteres»
Para abordar este enigma fundamental, la comunidad científica ha desarrollado varios marcos teóricos, de los cuales dos han sido particularmente influyentes: la hipótesis del «Mundo de ARN» y la teoría del «Mundo de Tioésteres».
La hipótesis del «Mundo de ARN» postula que, en una etapa temprana de la evolución de la vida, el ARN desempeñaba el papel central tanto en el almacenamiento de la información genética (una función ahora dominada por el ADN) como en la catálisis de reacciones bioquímicas (una función ahora dominada por las proteínas).2 Esta idea ganó un apoyo experimental masivo con el descubrimiento de los ribozimas, moléculas de ARN con actividad catalítica. Este hallazgo demostró que un único tipo de biopolímero podría, en principio, sostener un sistema biológico autosuficiente. En este escenario, un ribosoma primitivo compuesto enteramente de ARN podría haber catalizado la formación de los primeros péptidos. Sin embargo, la hipótesis del Mundo de ARN ha luchado por proporcionar un mecanismo químico detallado y robusto para el paso inicial y crucial: la aminoacilación del ARN en ausencia de las enzimas aaRS. ¿Cómo se «cargaron» los primeros ARNt con aminoácidos de manera eficiente y selectiva en un entorno acuoso?
En contraste, la teoría del «Mundo de Tioésteres» propone que la vida no comenzó con la replicación de polímeros informacionales, sino con un metabolismo primitivo impulsado por la química de compuestos de azufre de alta energía, específicamente los tioésteres.2 Un tioéster es un análogo de un éster en el que un átomo de azufre reemplaza a uno de los oxígenos. Los tioésteres, como el acetil-CoA, son intermediarios centrales en el metabolismo contemporáneo y poseen una alta energía libre de hidrólisis, lo que los convierte en excelentes agentes de acilación.6 La teoría sugiere que los tioésteres de aminoácidos (aminoacil-tioésteres) podrían haberse formado en la Tierra primitiva y haber impulsado la síntesis no codificada de péptidos, creando un repertorio de moléculas funcionales antes de la llegada de la genética basada en ácidos nucleicos.5 Esta hipótesis proporciona una fuente plausible de energía química y reactividad, pero carece del componente informacional inherente al Mundo de ARN. No explica cómo la síntesis de péptidos pasó de ser aleatoria a ser dirigida por una plantilla.
La Necesidad de un Puente Conceptual y Químico
Ambas hipótesis, aunque poderosas en sus respectivos dominios, dejan lagunas significativas. El Mundo de ARN tiene la información pero lucha con la química funcional inicial, mientras que el Mundo de Tioésteres tiene la química energética pero carece de un sistema de información. La frontera de la investigación sobre el origen de la vida ha sido, durante mucho tiempo, la búsqueda de un puente conceptual y, lo que es más importante, químico, entre estos dos escenarios. ¿Cómo podrían haberse integrado la reactividad de los tioésteres y la capacidad informacional del ARN para dar lugar al sistema de traducción codificada que vemos hoy?
El trabajo de Singh et al., publicado en Nature bajo el título «Aminoacilación de ARN mediada por tioésteres y síntesis de peptidil-ARN en agua», es de una importancia capital precisamente porque construye este puente.2 La investigación no busca reemplazar una hipótesis con la otra, sino que proporciona una vía química experimentalmente validada que demuestra cómo los intermediarios reactivos de un Mundo de Tioésteres podrían haber sido cooptados para funcionalizar los polímeros informacionales de un Mundo de ARN.6 Al hacerlo, el estudio ilumina un camino plausible para la coevolución de los péptidos y los ácidos nucleicos, sentando las bases químicas para el surgimiento del código genético y la síntesis de proteínas dirigida por plantilla. El artículo reframa el problema del origen de la traducción no como la necesidad de replicar el complejo sistema enzimático moderno, sino como la búsqueda de un sistema
químicamente predispuesto, donde las propiedades inherentes de las moléculas precursoras dictan el resultado deseado de manera espontánea y selectiva, sin la necesidad de catálisis enzimática.
La Química del Desafío: Aminoacilación en un Mundo Acuoso Prebiótico
Obstáculos Termodinámicos y Cinéticos
La síntesis de los intermediarios clave en la producción de proteínas, los aminoacil-ésteres (como los aminoacil-ARNt), se enfrenta a desafíos químicos formidables en el entorno acuoso que se presume fue la cuna de la vida. Estos obstáculos son tanto termodinámicos como cinéticos y han representado una barrera importante para el desarrollo de modelos prebióticos plausibles.
El principal obstáculo es la hidrólisis. La formación de un enlace éster (o tioéster) a partir de un ácido carboxílico (el aminoácido) y un alcohol (el grupo hidroxilo del ARN) o un tiol es una reacción de condensación, lo que significa que libera una molécula de agua.1 Según el principio de Le Châtelier, realizar una reacción de este tipo en un disolvente que es también el producto (agua) es extremadamente desfavorable. El equilibrio termodinámico en un entorno acuoso se inclina abrumadoramente hacia la reacción inversa: la hidrólisis, la ruptura del enlace éster por el agua.1 En la Tierra primitiva, cualquier aminoácido activado que se formara estaría en una carrera constante contra la destrucción por el vasto exceso de moléculas de agua circundantes, cuya concentración es de aproximadamente 55 M. Esto significa que la hidrólisis no solo es la vía termodinámicamente favorecida, sino también una trampa cinética masiva.
El segundo gran desafío es la falta de selectividad, que conduce a la polimerización aleatoria y a una miríada de reacciones secundarias. Los aminoácidos son moléculas bifuncionales: poseen un grupo amino nucleofílico y un grupo carboxilo que, una vez activado, se vuelve electrofílico.1 En ausencia de la protección y la canalización que proporcionan los sitios activos de las enzimas, un aminoácido activado en solución es tan propenso a reaccionar con el grupo amino de otra molécula de aminoácido como con cualquier otro nucleófilo.1 Este proceso, la auto-condensación, conduce a la formación incontrolada de péptidos de secuencia aleatoria.1 Si bien la formación de péptidos es, en última instancia, el objetivo, esta polimerización no dirigida crea un «ruido» químico que ahogaría cualquier señal informacional incipiente. Para que un sistema de traducción codificada pudiera surgir, la formación de péptidos dirigida por una plantilla de ARN debía superar de alguna manera esta tendencia inherente a la síntesis aleatoria.1 El sistema debía encontrar una manera de activar un aminoácido para la ligación al ARN sin que esa misma activación condujera a una polimerización descontrolada.
El Fracaso de los Métodos Químicos Previos
Dada la magnitud de estos desafíos, no es de extrañar que, antes de la investigación de Singh et al., no existieran métodos químicos capaces de aminoacilar eficaz y selectivamente los dioles 2′,3′ del ARN con una amplia gama de aminoácidos proteinogénicos en agua.1 Los enfoques de la química orgánica sintética clásica para formar ésteres superan estos problemas utilizando estrategias que son completamente inverosímiles en un contexto prebiótico. Por ejemplo, las reacciones se llevan a cabo en disolventes anhidros (libres de agua) para evitar la hidrólisis, y se utilizan «grupos protectores» para bloquear temporalmente la reactividad del grupo amino y prevenir la auto-condensación.8 Estas condiciones controladas de laboratorio están muy lejos de la «sopa» acuosa y químicamente diversa de la Tierra primitiva.
El problema central que los métodos anteriores no lograron resolver es el de la quimioselectividad. En una mezcla prebiótica que contiene agua, aminas, grupos hidroxilo y tioles, todos compitiendo como nucleófilos, ¿cómo se puede dirigir una reacción para formar un enlace aminoacil-ARN específico, en el hidroxilo 2′ o 3′ del último nucleótido, y no en otro lugar? La solución propuesta por Singh et al. es notable por su elegancia química. Se basa en el uso de un intermediario, el aminoacil-tioéster, que posee una reactividad «afinada». Este intermediario es lo suficientemente estable cinéticamente como para resistir la hidrólisis rápida y, crucialmente, es sorprendentemente poco reactivo hacia los nucleófilos de amina a pH neutro, lo que suprime la polimerización aleatoria.1 Sin embargo, conserva una reactividad suficiente para transferir eficientemente su grupo aminoacilo a los grupos hidroxilo más ácidos y accesibles del ARN, es decir, el diol 2′,3′ terminal. Esta reactividad diferencial es la clave para resolver el problema de la quimioselectividad. El tioéster actúa como una especie de «compuerta cinética» prebiótica, que impide las vías de reacción no deseadas (hidrólisis, polimerización) mientras permite que proceda la reacción deseada (aminoacilación del ARN). Esto representa una forma de control químico que precede y presagia el control biológico mediado por enzimas.
Una Solución Plausible: La Vía de los Tioésteres de Aminoacilo
El trabajo de Singh et al. presenta una vía química de dos pasos, robusta y prebióticamente plausible, que supera los obstáculos descritos anteriormente. El primer paso es la formación de los intermediarios clave, los tioésteres de aminoacilo, a partir de precursores simples. El segundo paso es la transferencia selectiva del grupo aminoacilo desde el tioéster al ARN.
Formación de Intermediarios Clave: Síntesis de Tioésteres de Aminoacilo
El éxito de cualquier esquema prebiótico depende de la plausibilidad de sus materiales de partida. Los autores demuestran que los tioésteres de aminoacilo (estructuras generales 5 en su trabajo) pueden sintetizarse a partir de varias clases de precursores considerados relevantes para la química prebiótica: los aminonitrilos (2), los N-carboxianhídridos de aminoácidos (NCAs, 33) y los aminoacil-adenilatos (32).1 Estos precursores reaccionan con una variedad de tioles simples (
3), incluidos análogos de cofactores biológicos como la panteteína (3a) y el coenzima M (3c), para producir selectivamente los correspondientes aminoacil-tioésteres.1
La reacción de formación de tioésteres a partir de aminonitrilos y tioles demostró ser más eficiente en condiciones ligeramente ácidas, con un pH óptimo entre 3 y 5.1 A primera vista, esto podría parecer un obstáculo, ya que se cree que los océanos primitivos eran neutros o ligeramente alcalinos. Sin embargo, los investigadores propusieron y demostraron un mecanismo geoquímico ingenioso para generar estas condiciones localmente. Mostraron que la congelación de soluciones acuosas de fosfato de sodio, un mineral plausible en la Tierra primitiva, conduce a la formación de fases eutécticas donde el pH desciende de forma natural a aproximadamente 3.5.1 En estas condiciones de congelación, la reacción para formar tioéster de alanina (
5bAla) a partir de nitrilo de alanina (2Ala) y un tiol procedió de manera eficiente, alcanzando un rendimiento del 48% en 30 días, incluso partiendo de concentraciones de reactivos muy diluidas (1 mM).1 El proceso de congelación tiene el doble beneficio de crear el entorno de pH ácido necesario y de concentrar los reactivos, superando así el problema de la dilución en un océano primitivo.
Esta primera etapa del proceso es fundamental porque resuelve uno de los problemas clave de la química prebiótica de péptidos. La formación del intermediario tioéster actúa como un mecanismo de «activación controlada». A diferencia de otros intermediarios altamente reactivos como los NCAs, que tienden a polimerizar de forma explosiva en agua, los tioésteres son lo suficientemente estables como para acumularse y persistir, pero lo suficientemente activados para la siguiente etapa. La adición de un tiol a una solución de NCA desvía drásticamente la vía de reacción de la polimerización a la formación del tioéster.1 Por lo tanto, el tiol funciona como un «interruptor catalítico» que redirige el flujo químico de una vía destructiva y aleatoria a una vía constructiva y específica.
La Carga del ARN: Transferencia Selectiva del Aminoácido
Una vez formados, los aminoacil-tioésteres (5) demuestran ser agentes de acilación notablemente eficaces y selectivos para el ARN en condiciones acuosas a pH neutro.2 Cuando se incuban con ribonucleósidos libres (como la uridina,
17U) o con oligómeros de ARN, el grupo aminoacilo se transfiere desde el átomo de azufre del tioéster a uno de los grupos hidroxilo del azúcar de ribosa, formando un aminoacil-ARN.1
La característica más llamativa de esta reacción es su alta selectividad por el diol vecinal 2′,3′ del anillo de ribosa.1 Químicamente, esta selectividad puede explicarse por la mayor acidez (menor
pKa) de estos grupos hidroxilo en comparación con otros alcoholes en la molécula, como el hidroxilo 5′. Los autores confirmaron esta selectividad mediante una serie de experimentos de control. Los 2′-desoxinucleósidos (que carecen del hidroxilo 2′) mostraron rendimientos de aminoacilación muy bajos, mientras que los nucleósidos modificados que poseían un hidroxilo 2′ o 3′ aislado (como la 3′-desoxiadenosina 20A o la 2′-O-metoxi-adenosina 23A) fueron acilados de manera sustancial.1 La supresión casi total de la reacción con 2′,3′-didesoxiadenosina (
22A), que carece de ambos hidroxilos, confirmó que el diol 2′,3′ es el sitio de reacción principal.1
Un detalle mecanicista sutil pero crucial es el papel del grupo amino libre del propio aminoácido. Los experimentos demostraron que un tioéster de alanina acetilado (Ac-Ala-SEt), que tiene su grupo amino bloqueado, era completamente incapaz de reaccionar con el diol del ARN.1 Esto sugiere fuertemente que el grupo amino participa en la reacción, probablemente actuando como un catalizador general de base intramolecular que desprotona el hidroxilo del ARN, facilitando su ataque nucleofílico sobre el carbono del tioéster. Esta auto-catálisis es un ejemplo elegante de cómo las propiedades inherentes de los bloques de construcción de la vida pueden guiar las reacciones químicas por vías productivas.
La siguiente tabla resume los datos cuantitativos clave de la vía de dos pasos, ilustrando la robustez y generalidad de la química.
| Tabla 1: Eficiencia en la Síntesis de Tioésteres de Aminoacilo y Aminoacilación de ARN | ||||
| Parte A: Síntesis de Tioésteres de Aminoacilo a partir de Aminonitrilos | ||||
| Aminoácido | Tiol | Condiciones | Rendimiento (%) | Referencia |
| Alanina | 3-mercaptopropiónico | pH 3-5, T ambiente | 13-48 | 1 |
| Alanina | 3-mercaptopropiónico | pH 3.5 (congelación, -7 °C), 30 días | 48 | 1 |
| Glicina | 3-mercaptopropiónico | pH 3.5 (congelación) | 46 | 1 |
| Leucina | 3-mercaptopropiónico | pH 3.5 (congelación) | 64 | 1 |
| Arginina | 3-mercaptopropiónico | pH 3.5 (congelación) | 41 | 1 |
| Lisina | 3-mercaptopropiónico | pH 3.5 (congelación) | 44 | 1 |
| Parte B: Aminoacilación de ARN mediada por Tioésteres | ||||
| Tioéster | Sustrato de ARN | Condiciones | Rendimiento (%) | Referencia |
| Alanyl-tioéster | Uridina (17U) | pH 6.5, 600 mM tioéster | 50 | 1 |
| Alanyl-tioéster | Uridina (17U) | pH 6.5 (congelación, -7 °C), 10 mM tioéster | 58 | 1 |
| Alanyl-tioéster | 5′-mercapto-adenosina | pH 6.5, T ambiente | 54 (en diol 2′,3′) | 1 |
| Alanyl-tioéster | ds-ARN (ON4) | pH 6.5, T ambiente | ~30 | 1 |
| Arginyl-tioéster | ds-ARN (ON4) | pH 6.5, T ambiente | 64 | 1 |
El Papel de la Estructura: La Directiva del Dúplex de ARN Nativo
Quizás el descubrimiento más profundo y revelador del estudio de Singh et al. es el papel crítico que juega la estructura tridimensional del ARN en la dirección de la reactividad química. Demostraron que la formación de una doble hélice de ARN transforma una reacción de aminoacilación no específica en un proceso altamente selectivo, proporcionando una justificación química fundamental para la evolución de estructuras de ARN complejas como el ARNt.
ARN Monocatenario (ssRNA): Aminoacilación No Específica
Como experimento de control crucial, los investigadores primero examinaron la reacción del tioéster de alanina con un oligómero de ARN de cadena sencilla (ssRNA). El resultado fue claro: la aminoacilación se produjo de manera indiscriminada a lo largo de la cadena de ARN.1 Además de la reacción esperada en el diol 2′,3′ del extremo 3′ terminal, se observaron modificaciones significativas en numerosos grupos hidroxilo 2′ internos de los residuos de ribonucleótidos a lo largo de la hebra.
Este resultado es de vital importancia porque demuestra que la quimioselectividad inherente del tioéster por los grupos hidroxilo de la ribosa no es suficiente por sí sola para lograr la especificidad de sitio necesaria para un sistema de traducción funcional. En la biología moderna, el aminoácido siempre se añade al extremo 3′ del ARNt, el «extremo comercial» de la molécula que interactúa con el centro peptidil transferasa del ribosoma. Una modificación aleatoria a lo largo de la cadena sería un callejón sin salida evolutivo. Para que la síntesis de péptidos dirigida por plantilla pudiera comenzar, era necesario un mecanismo que asegurara que la aminoacilación ocurriera exclusivamente en la posición correcta.
ARN Bicatenario (dsRNA): Resurrección de la Selectividad
El avance conceptual se produjo cuando el equipo repitió el experimento, pero esta vez utilizando un ARN de doble cadena (dsRNA), formado al hibridar el oligómero de ARN original con su hebra complementaria.2 El resultado fue una transformación espectacular en el perfil de reactividad. La formación del dúplex de ARN suprimió drásticamente la aminoacilación de los grupos hidroxilo 2′ internos, y la reacción se dirigió casi exclusivamente al diol 2′,3′ del nucleótido terminal 3′.1
La explicación de este fenómeno radica en la estructura de la doble hélice de ARN de tipo A. En esta conformación, las bases de nucleótidos están orientadas hacia el interior de la hélice, y los grupos hidroxilo 2′ de la columna vertebral de azúcar-fosfato quedan relativamente enterrados y estéricamente inaccesibles dentro del surco menor de la hélice.1 Por el contrario, el extremo 3′ de una hebra de ARN suele ser más flexible y expuesto al disolvente. Por lo tanto, la formación del dúplex actúa como una «máscara» o «grupo protector» físico y autoensamblado. No requiere catálisis; es una consecuencia inherente de las reglas de apareamiento de bases de Watson-Crick. Esta estructura protege los sitios internos no deseados de la reacción y deja el sitio terminal 3′ deseado como el único objetivo accesible para el aminoacil-tioéster.
Este hallazgo representa una forma primitiva pero potente de transferencia de información, donde la información estructural (la conformación de la doble hélice) se traduce directamente en información química (la ubicación específica de un enlace covalente). Demuestra que los principios de la biología molecular, como la relación entre estructura y función, pueden operar a un nivel puramente químico y prebiótico. Las mismas reglas de apareamiento de bases que son fundamentales para el almacenamiento y la replicación de la información genética también desempeñaron un papel crucial y no genético en el control de la reactividad química, mucho antes de la evolución de los sitios activos de las enzimas.
Implicaciones para la Evolución del ARNt
Las implicaciones de este descubrimiento para nuestra comprensión de la evolución del ARNt son profundas. La estructura de hoja de trébol canónica de un ARNt moderno presenta varias regiones de doble hélice, incluido el «tallo aceptor», que es el dúplex que culmina en el extremo 3′ donde se une el aminoácido. El hallazgo de Singh et al. proporciona la primera razón química convincente de por qué evolucionó esta estructura. El tallo aceptor no es simplemente un andamio estructural; es una herramienta química pasiva, un «proto-enzima», que utiliza su estructura para dirigir la aminoacilación al sitio correcto.
Este modelo sugiere una coevolución gradual de la estructura del ARN y la función química. Las primeras moléculas de ARN que adoptaron conformaciones de dúplex estables habrían sido aminoaciladas de forma más selectiva y, por lo tanto, habrían sido precursoras más eficaces para la síntesis de péptidos. Esto habría creado una presión selectiva para la evolución de estructuras de ARN cada vez más complejas y estables, capaces de ejercer un control cada vez mayor sobre las reacciones químicas. El dúplex de ARN no es solo una plantilla de información; es una herramienta funcional que surgió de las propiedades fisicoquímicas inherentes del propio polímero.
La siguiente tabla ilustra cuantitativamente el dramático efecto de la estructura del ARN en la selectividad de la reacción.
| Tabla 2: Comparación de la Selectividad de Aminoacilación en ARN monocatenario (ssRNA) vs. bicatenario (dsRNA) | |||
| Sustrato de ARN | Estructura | Rendimiento Total de Aminoacilación (%) | Distribución de la Aminoacilación |
| Oligonucleótido ON4 | Monocatenario (ssRNA) | Elevado | No específica: Múltiples sitios internos + 3′-terminal |
| Oligonucleótido ON4 | Bicatenario (dsRNA) | Moderado a alto (~30%) | Altamente selectiva: Casi exclusivamente en el diol 2′,3′ del 3′-terminal |
Nota: Los valores porcentuales exactos para la distribución no se especifican numéricamente en los resúmenes, pero la descripción cualitativa de «extensa y no específica» versus «selectiva» es un hallazgo central.1
Alcance y Versatilidad de la Reacción
Un requisito fundamental para cualquier vía química propuesta para el origen de la vida es que debe ser lo suficientemente general como para dar cuenta de la diversidad de los bloques de construcción biológicos. El sistema de aminoacilación mediado por tioésteres de Singh et al. demuestra una notable versatilidad, funcionando eficazmente con una amplia gama de cadenas laterales de aminoácidos y revelando comportamientos químicos complejos que insinúan los orígenes de la catálisis.
Amplia Tolerancia de Cadenas Laterales de Aminoácidos
La investigación demostró que tanto la formación de tioésteres como la posterior aminoacilación del ARN son compatibles con una gran diversidad de aminoácidos proteinogénicos.1 El estudio informa del éxito de la reacción con residuos que abarcan todo el espectro de propiedades químicas:
- No polares (hidrofóbicos): Alanina (Ala), Valina (Val), Leucina (Leu), Fenilalanina (Phe), Prolina (Pro).
- Polares no cargados: Serina (Ser), Glicina (Gly), Glutamina (Gln).
- Ácidos (cargados negativamente): Aspartato (Asp), Glutamato (Glu).
- Básicos (cargados positivamente): Arginina (Arg), Lisina (Lys).
- Aromáticos: Fenilalanina (Phe), Histidina (His).
Esta amplia tolerancia es de una importancia crítica. Sugiere que la química fundamental no está restringida a unos pocos aminoácidos simples, sino que es lo suficientemente robusta como para haber podido generar un conjunto diverso de aminoacil-ARNs en la Tierra primitiva. Esto proporciona una base química para la eventual selección del conjunto canónico de 20 aminoácidos utilizados por la vida. La capacidad de cargar ARN con aminoácidos funcionalmente diversos (ácidos, básicos, hidrofóbicos) es un requisito previo para la síntesis de péptidos con capacidades estructurales y catalíticas.
Casos Especiales y Catálisis Intramolecular
Más allá de la amplia generalidad, el estudio descubrió casos específicos de reactividad que son particularmente reveladores y sugieren la aparición de un comportamiento químico más sofisticado.
Uno de los ejemplos más notables es la reactividad del aspartato. El aspartato es un aminoácido dicarboxílico, lo que significa que tiene dos grupos carboxilo: el α-carboxilo que forma parte de la cadena principal del péptido y el β-carboxilo en su cadena lateral. La activación del aspartato (por ejemplo, como Asp-NCA, 33Asp) podría, en principio, conducir a la formación de enlaces a través de cualquiera de los dos grupos. Sin embargo, los investigadores encontraron que la reacción con tioles procedía con una alta selectividad cinética para formar el α-aspartil-tioéster (5cAsp) sobre el isómero β (con una relación de 11:1).1 Aún más importante, esta selectividad por el enlace
α se mantuvo durante la posterior transferencia al ARN.1 Este es un hallazgo crucial, ya que demuestra un sesgo químico inherente hacia la formación de la conectividad «correcta» de la cadena principal del péptido, incluso en ausencia de una enzima que dirija la reacción.
Quizás el resultado más sorprendente fue el observado con la arginina. La aminoacilación de un dsRNA con el tioéster de arginina (L-5eArg) se produjo con un rendimiento excepcionalmente alto del 64%, significativamente mayor que el de muchos otros aminoácidos.1 Los autores atribuyen esta eficiencia mejorada a un efecto de catálisis intramolecular. La cadena lateral de la arginina contiene un grupo guanidinio, que está cargado positivamente a pH neutro. Esta carga positiva puede formar fuertes interacciones electrostáticas y de enlace de hidrógeno con la columna vertebral de fosfato cargada negativamente del ARN. Esta interacción probablemente pre-organiza y ancla el tioéster de arginina cerca del extremo 3′ del ARN, posicionándolo de manera óptima para la reacción. Esta pre-organización reduce la barrera de activación entrópica de la reacción, lo que constituye una forma de catálisis.
Este hallazgo es un ejemplo asombroso de un monómero individual que cataliza su propia ligación a un polímero. Es un presagio de la función enzimática y sugiere que los aminoácidos canónicos podrían no haber sido seleccionados al azar. Es posible que fueran seleccionados, en parte, por sus propiedades químicas intrínsecas que facilitaban las mismas reacciones necesarias para construir los primeros biopolímeros funcionales. Esto apunta a un posible bucle de retroalimentación en la evolución química: los monómeros que son intrínsecamente «mejores» para incorporarse a los polímeros tendrían una mayor probabilidad de formar parte de los primeros péptidos funcionales. Si esos péptidos confirieran una ventaja (por ejemplo, estabilizando el ARN), se crearía una presión selectiva para desarrollar un sistema que codificara específicamente esos aminoácidos útiles, lo que podría haber sentado las bases para el origen del código genético.
Hacia la Síntesis de Péptidos: Formación del Enlace Peptídico
El objetivo final de la aminoacilación del ARN es proporcionar los sustratos para la síntesis de péptidos. Un aspecto crucial del trabajo del equipo de Powner, destacado en los informes de prensa que acompañan a la publicación principal, es que demostraron no solo la carga del ARN, sino también el siguiente paso: la formación de un enlace peptídico.2
De la Aminoacilación a la Peptidilación
Una vez que se ha formado un aminoacil-ARN, este se convierte en el sustrato para la elongación de la cadena peptídica. Los investigadores demostraron que una molécula de aminoacil-ARN, preparada mediante su método mediado por tioésteres, podía reaccionar con un segundo aminoácido activado (por ejemplo, en forma de un aminotioácido) para formar un dipeptidil-ARN.2 En esta reacción, el grupo amino del segundo aminoácido ataca el carbono del carbonilo del enlace éster del primer aminoacil-ARN, formando un enlace peptídico y transfiriendo el primer aminoácido al segundo. El resultado es una molécula de ARN que ahora lleva un dipéptido en su extremo 3′.
Esta demostración es la culminación de la vía química propuesta. Valida que el producto de la etapa de aminoacilación no es un callejón sin salida químico, sino un intermediario productivo y viable que está preparado para la siguiente reacción en la secuencia de síntesis de proteínas. La química es un eco directo de la reacción fundamental que tiene lugar en el Centro Peptidil Transferasa (PTC) del ribosoma moderno, donde el grupo amino de un aminoacil-ARNt en el sitio A ataca el enlace éster de un peptidil-ARNt en el sitio P.7 El hecho de que esta química central pueda ocurrir en agua sin la ayuda de la masiva maquinaria ribosómica sugiere que la reacción de formación de enlaces peptídicos en un andamio de ARN es un proceso químico fundamentalmente predispuesto.
Demostración de un Ciclo Completo de Elongación
Al demostrar tanto la aminoacilación como la posterior formación del enlace peptídico, el estudio establece un ciclo completo y no enzimático para una ronda de elongación de la cadena peptídica. La secuencia de reacciones se puede resumir de la siguiente manera:
- Activación: Un aminoácido precursor (como un aminonitrilo) reacciona con un tiol para formar un aminoacil-tioéster, un intermediario activado y relativamente estable.
- Carga: El aminoacil-tioéster reacciona con el extremo 3′ de una molécula de ARN (preferentemente en una estructura de dúplex) para formar un aminoacil-ARN.
- Elongación: El aminoacil-ARN reacciona con un segundo aminoácido activado para formar un dipeptidil-ARN.
Este ciclo completo proporciona una base química plausible para un proto-ribosoma. En este escenario primitivo, una estructura de ARN más grande podría haber actuado como un andamio para unir y orientar una molécula de peptidil-ARN y una molécula de aminoacil-ARN entrante, facilitando la reacción de transferencia de péptidos y permitiendo la adición secuencial de aminoácidos. Es importante destacar que la energía necesaria para la formación del enlace peptídico (que es termodinámicamente desfavorable por sí misma) se «carga» en el sistema durante la etapa inicial de activación para formar el tioéster o el posterior aminoacil-éster. Las etapas posteriores son transferencias de acilo energéticamente cuesta abajo. Este principio de activación en dos etapas (primero la activación del monómero, luego la polimerización) es un tema central en toda la biosíntesis y se demuestra aquí que es químicamente factible en un contexto prebiótico.
Implicaciones y Perspectivas Futuras: Uniendo los Mundos del ARN y los Tioésteres
Una Síntesis de Hipótesis y un Nuevo Paradigma
El verdadero poder del trabajo de Singh et al. no reside simplemente en el descubrimiento de una nueva serie de reacciones, sino en su capacidad para sintetizar y unificar ideas previamente dispares sobre el origen de la vida. En lugar de apoyar la hipótesis del Mundo de ARN en detrimento de la del Mundo de Tioésteres, o viceversa, la investigación las fusiona elegantemente en un único marco coherente y químicamente plausible.2
Demuestra cómo la química energética del Mundo de Tioésteres (la formación de intermediarios de tioéster de alta energía) podría haber proporcionado la «carga» funcional para los polímeros informacionales del Mundo de ARN. Este acoplamiento habría creado los primeros péptidos funcionales de una manera dirigida por la estructura del ARN, iniciando un proceso de coevolución entre los dos tipos de biopolímeros que finalmente condujo al sistema de traducción codificada. Este nuevo paradigma sugiere que la vida no surgió de un «mundo» de un solo polímero, sino de la interacción sinérgica entre diferentes clases de moléculas, cada una aportando propiedades únicas (información, catálisis, energía) al sistema emergente.
Preguntas Abiertas y Futuras Direcciones de Investigación
Como toda investigación fundamental, este trabajo abre más preguntas de las que responde, trazando el rumbo para futuras exploraciones en el campo del origen de la vida.
La pregunta más importante y desafiante que queda es el origen de la especificidad de secuencia, es decir, el código genético.2 El sistema actual demuestra la
química de la aminoacilación y la síntesis de péptidos, pero no la información. No explica cómo secuencias de ARN específicas (proto-codones en un ARNm y proto-anticodones en un ARNt) se asociaron de manera única con aminoácidos específicos. ¿Existen afinidades estereoquímicas directas entre ciertos aminoácidos y sus codones o anticodones correspondientes? ¿O el código surgió a través de un proceso de «accidente congelado», donde las asignaciones iniciales, aunque arbitrarias, se fijaron evolutivamente? La plataforma química desarrollada por Singh et al. proporciona ahora una herramienta para probar experimentalmente estas hipótesis.
Otra cuestión crítica es el origen de la homoquiralidad biológica. La vida utiliza casi exclusivamente L-aminoácidos y D-azúcares. El estudio actual observó solo una diastereoselectividad menor en la reacción de aminoacilación, con una ligera preferencia por un diastereómero sobre otro cuando el ARN terminaba en una purina.1 Comprender cómo esta pequeña preferencia inicial podría haberse amplificado para dar lugar a la homoquiralidad absoluta que vemos hoy es un área clave para la investigación futura.
Finalmente, queda la cuestión de la transición de la catálisis pasiva dirigida por la estructura a la catálisis activa mediada por ribozimas. El estudio ofrece una pista tentadora: observaron que un dúplex de ARN con un desajuste en el extremo 3′ (un dúplex imperfecto) en realidad mejoraba la velocidad y el rendimiento de la aminoacilación, incluso a concentraciones muy bajas de tioéster.1 Esto sugiere que las estructuras de ARN «desordenadas» o no canónicas podrían haber poseído propiedades catalíticas incipientes, actuando como los primeros y rudimentarios sitios activos. Explorar cómo la evolución de tales estructuras podría haber conducido a un proto-ribosoma con una actividad peptidil transferasa mejorada será fundamental para completar la historia del origen de la traducción.
Conclusión Final: Un Paso Plausible en el Camino Hacia la Vida
El origen de la vida sigue siendo uno de los problemas más complejos y multifacéticos de la ciencia. Sin embargo, la investigación sobre la aminoacilación de ARN mediada por tioésteres representa un avance conceptual y experimental de primer orden. Transforma un paso clave en el origen de la síntesis de proteínas, que antes era un problema en gran medida especulativo, en un sistema químico tratable y bien definido. El trabajo de Singh et al. proporciona una de las vías químicas más completas, plausibles y experimentalmente robustas hasta la fecha para explicar cómo la unión de la química del azufre y la biología del ARN podría haber sentado las bases para la aparición de la vida tal como la conocemos. Si bien el viaje desde la química prebiótica hasta la primera célula viva fue sin duda largo y tortuoso, este estudio ilumina un tramo crucial de ese camino con una claridad sin precedentes.
Obras citadas
- Thioester-mediated chemoselective aminoacylation of … – ChemRxiv, fecha de acceso: agosto 27, 2025, https://chemrxiv.org/engage/api-gateway/chemrxiv/assets/orp/resource/item/66bddb21f3f4b05290f1dc7e/original/Aminoacylation_Manuscript_preprint.pdf
- RNA and Sulfur Compounds Possibly Created the First Peptides on Earth | The Scientist, fecha de acceso: agosto 27, 2025, https://www.the-scientist.com/rna-and-sulfur-compounds-possibly-created-the-first-peptides-on-earth-73323
- Synthesis of Peptidyl-tRNA Mimics for Structural Biology Applications – PubMed Central, fecha de acceso: agosto 27, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC10552525/
- A Flexible, Scalable Method for Preparation of Homogeneous Aminoacylated tRNAs – PMC, fecha de acceso: agosto 27, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC4267853/
- Aminoacyl-tRNA synthetases and the evolution of coded peptide synthesis: the Thioester World – PubMed, fecha de acceso: agosto 27, 2025, https://pubmed.ncbi.nlm.nih.gov/26831912/
- Thioesters Support Efficient Protein Biosynthesis by the Ribosome | ACS Central Science, fecha de acceso: agosto 27, 2025, https://pubs.acs.org/doi/10.1021/acscentsci.4c01698
- Thioesters Support Efficient Protein Biosynthesis by the Ribosome – PMC – PubMed Central, fecha de acceso: agosto 27, 2025, https://pmc.ncbi.nlm.nih.gov/articles/PMC11950863/
- ChemInform Abstract: Direct Thioesterification from Carboxylic Acids and Thiols Catalyzed by a Broensted Acid. – ResearchGate, fecha de acceso: agosto 27, 2025, https://www.researchgate.net/publication/11259205_ChemInform_Abstract_Direct_Thioesterification_from_Carboxylic_Acids_and_Thiols_Catalyzed_by_a_Broensted_Acid
- (PDF) Thioester-mediated chemoselective aminoacylation of RNA in water. – ResearchGate, fecha de acceso: agosto 27, 2025, https://www.researchgate.net/publication/383185444_Thioester-mediated_chemoselective_aminoacylation_of_RNA_in_water
- Thioester-mediated chemoselective aminoacylation of RNA in water. – ChemRxiv, fecha de acceso: agosto 27, 2025, https://chemrxiv.org/engage/chemrxiv/article-details/66bddb21f3f4b05290f1dc7e